-
Email info@annpnc.org
-
Address 848 N. Rainbow Blvd. #5486 Las Vegas, NV 89107, USA
1Third Year, Senior Resident, Department of Radiodiagnosis, KGMU, Lucknow, UP, India.
2Additional Professor, Department of Radiodiagnosis, KGMU, Lucknow, UP, India.
3Professor and Head, Department of Radiodiagnosis, KGMU, Lucknow, UP, India
*Corresponding author: Shubhi Agarwal
Third Year, Senior Resident, Deptartment of Radiodiagnosis,
KGMU, Lucknow, UP, India.
Email ID: shubhi93agarwal@gmail.com
Received: Apr 07, 2025
Accepted: May 15, 2025
Published Online: May 22, 2025
Journal: Annals of Pediatrics and Neonatal Care
Copyright: Agarwal S et al. © All rights are reserved
Citation: Agarwal S, Srishti S, Sukriti K, Anit P. Navigating the genetic landscape of failure to thrive in infancy: Glycosylphosphatidylinositol biosynthesis defect chronicles. Ann Pediatr Neonatal Care. 2025; 1(1): 1008.
Glycosylphosphatidylinositol is a glycolipid that anchors proteins to the cell membrane, synthesis of (GPI-APs) is important for protein processing and function like embryogenesis, immune response, neurogenesis.
GPI synthesis and modification are mediated by at least 29 genes and loss-of-function in 19 of these genes have been described to cause neurological impairments, intellectual disability, Developmental Delay (DD), and multiple congenital anomalies and dysmorphisms.
Here, we report a case of mutation in PIGW gene in a 5-month-old female child with recurrent pneumonia, failure to thrive, neurological and dermatological manifestations.
A 5-month-old female child born out of non-consanguineous parents presented to us with:
◆ Failure to thrive, developmental delays, recurrent pneumonia.
◆ On and off fever with respiratory difficulty since birth.
◆ There were no neonatal risk factors, few episodes of seizures were present, the infant was partially immunized, On Clinical examination there were hypopigmented patches over the abdomen with coarse hair and alopecia.
Biochemical findings revealed raised ammonia levels with decreased hemoglobin with normal ALP levels.
EEG revealed hypsarrythmia.
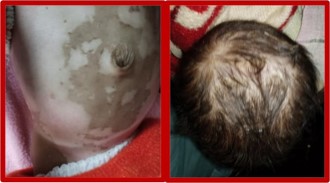
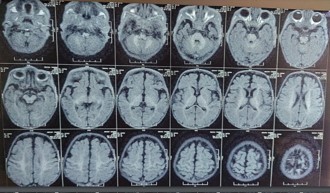
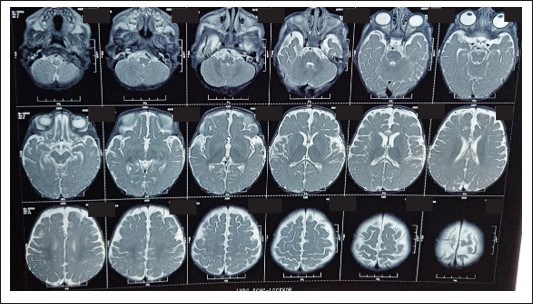
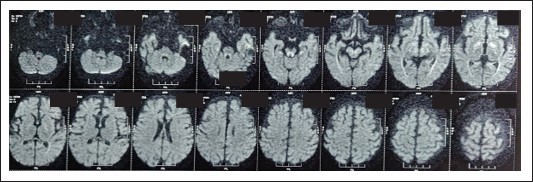
Radiological findings
HRCT revealed consolidations in bilateral upper and lower lung lobes with mild hepatosplenomegaly and borderline cardiomegaly MRI Brain revealed: cerebral atrophy, thinning of corpus callosum and symmetric DWI restriction in globus pallidi, PLIC, peritrigonal white matter, optic radiations, brainstem and medial temporal gyri (which raised the suspicion of INBORN ERROR OF METABOLISM).
Radiological differential of Congenital biotinidase/holocarboxylase synthetase deficiency were being considered in view of clinico biochemical finding correlation.
Whole exome genome sequencing revealed PIGW gene defect (glycosylphosphatidylinositol biosynthesis defect 11) with homozygous variant and autosomal recessive inheritance.
GPI is synthesised in a multistage pathway, which involves the products of 23 genes.
Mutations in these genes lead to severe congenital syndromes known as GPI biosynthesis disorders (GPIBDs). Their phenotype consists of developmental delays (both motor and intellectual), encephalopathies, muscular hypotonia, a high prevalence of epileptic seizures, facial dysmorphisms, and cerebellar dysfunction.
Only six patients with homozygous or compound heterozygous pathogenic variants of PIGW have been identified in the literature thus far (our case had a homozygous autosomal recessive variant). Epileptic seizures were reported in all patients, and the most common types of seizures were epileptic spasms.
Distinctive facial and physical features and recurrent respiratory infections are common in these patients with developmental delays, as in our case which primarily presented with recurrent pneumonia and on and off seizures.
PIGW-related glycosylphosphatidylinositol deficiency is characterized by developmental delay, epilepsy, distinctive facial features, and multiple organ anomalies. Genetic testing is an important method for diagnosing this disease.
Flow cytometry and serum ALP level detection are crucial complements for genetic testing.
