-
Email info@annpnc.org
-
Address 848 N. Rainbow Blvd. #5486 Las Vegas, NV 89107, USA
1Division of Neonatology, Carlo Poma General Hospital, ASST Mantova, Italy.
2Division of Pediatrics, S. Chiara General Hospital, Azienda Provinciale per i Servizi Sanitari, Trento, Italy.
3Genetic Unit, Azienda Ospedaliera Universitaria Integrata of Verona, Verona, Italy.
4Division of Metabolic Diseases, Department of Medicine, University Hospital of Padova, Padova, Italy.
5Department of Pediatrics, Regional Centre for Newborn Screening, Diagnosis and Treatment of Inherited Metabolic Diseases and Congenital Endocrine Diseases, Azienda Ospedaliera Universitaria Integrata of Verona, Verona, Italy.
6Division of Cardiology, Department of Medicine, University of Verona, Verona, Italy.
7Centre for Medical Sciences (Cismed), University of Trento, Trento, Italy.
8Metabolic and Syndromic Disease Unit, Regional Coordinating Centre for Rare Diseases Udine, Azienda Sanitaria Universitaria Friuli Centrale, Italy.
#These authors have been equally contributed to this article.
*Corresponding author: Evelina Maines
Pediatric Unit, “S. Chiara” Hospital, Largo Medaglie d’oro, 9,
38122 Trento, Italy.
Email ID: evelina.maines@apss.tn.it
Tel: +39 0461 903538, Fax: +39 0461 903824
Received: Apr 15, 2025
Accepted: May 13, 2025
Published Online: May 20, 2025
Journal: Annals of Pediatrics and Neonatal Care
Copyright: Maines E et al. © All rights are reserved.
Citation: Morandi G, Maines E, Rodella G, Gugelmo G, Vitturi N, et al. Early cardiac involvement in MELAS syndrome: A pediatric case report. Ann Pediatr Neonatal Care. 2025; 1(1): 1007.
Keywords: MELAS; Mitochondrial; Pediatric; Cardiomyopathy.
A 1-year-old boy with a history of hypotonia and recurrent episodes of lactic acidosis during seasonal illnesses, was diag- nosed with MELAS (Mitochondrial encephalopathy lactic acido- sis with Stroke-like episodes) syndrome, following genetic con- firmation of a maternally inherited 3243A>G mutation in mito- chondrial DNA (mtDNA).
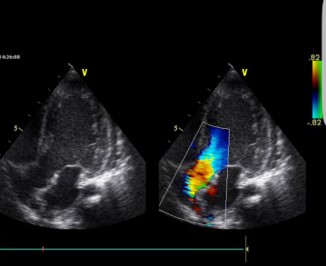
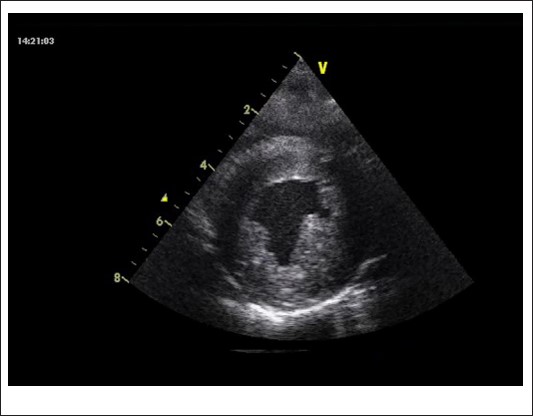
At 2 years of age, a gallop rhythm was detected on cardiac auscultation, raising concerns about underlying cardiomyopathy (CM). Echocardiography revealed hypertrophic cardiomyopathy (HCM) with diastolic dysfunction and mitral valve regurgitation (E/E’ 12.5, LVEDD 32 mm, z-score +1,24; LVESD 15 mm, z-score 1,20, IVS 6 mm, z-score +1,30, LVPW 6 mm, z-score +2,26, LVFS 53%, LVEDV 56 ml/m2, LVESV 26 ml/m2, LVEF 54%) (Figures 1a & 1b).
Given these findings, the patient was started on Carvedilol (1 mg/kg/day) and Aldactazide (0.3 mg/kg/day) to manage car- diac function and reduce preload.
Beyond cardiac involvement, the child exhibited poor growth and feeding difficulties due to early fatigue, a slow eating pace, and poor appetite. He required enteral feeding with a standard semi-elemental formula enriched with medium-chain triglycer- ides, by initially via nasogastric tube and later via percutaneous endoscopic gastrostomy. Attempts to increase feeding volumes or use concentrated/hypercaloric formulas resulted in gastro- intestinal intolerance. Despite nutritional support, growth re- mained unsatisfactory, with weight and height z-scores of -2.
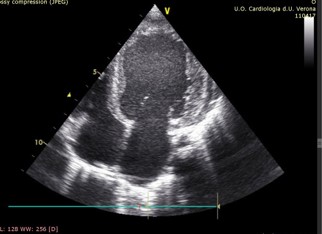
By age 4, progression to dilated cardiomyopathy (DCM) with further worsening of diastolic function was observed (LVEDV 84 ml/m2, LVESV 52 ml/m2, LVEF 38%) (Figure 2). The patient also exhibited psychomotor delay and moderate neurologic dysfunction - hallmark features of MELAS syndrome in children.
Clinical and instrumental evaluation for heart transplanta- tion (Htx) was being planned, despite significant ethical con- cerns due to the severity of multisystemic involvement, includ- ing neurologic impairment. The patient died due to sudden and severe cardiac dysfunction at the age of 4.6 years.
MELAS syndrome is one of the most common mitochondrial disorders, affecting approximately 1 in 4,000 individuals [1,2]. It is caused by pathogenic mutations in mtDNA, with the single-nucleotide variation m.3243A>G accounting for approximately 80% of cases [1,2]. The m.3243A>G mutation disrupts mitochondrial protein synthesis and electron transport chain function, leading to impaired oxidative phosphorylation(OXPHOS). As a result, mitochondrial energy production becomes insufficient to meet the high metabolic demands of various organs, especially during periods of metabolic stress [1,2]. Organs with high-energy requirements, such as the brain, eyes, heart and skeletal muscle, are the most severely affected in MELAS syndrome. Energy deficiency can also contribute to vascular endothelial dysfunction resulting in angiopathy and reduced blood perfusion in the microvasculature of several organs. These processes contribute to the stroke-like episodes characteristic of the disease [1,2].
MELAS syndrome is characterized by extreme variability in clinical presentation among affected individuals, even within the same family. This is largely due to heteroplasmy, the phe- nomenon in which different proportions of mutant and wild- type mtDNA coexist within different tissues of the same indi- vidual [2,3].
MELAS syndrome usually presents during childhood but adult-onset is possible. Children typically display a broad, multi-systemic phenotype with predominantly neurological manifes- tations. In older patients, onset commonly involves recurrent episodes of migraine-like headaches, seizures, encephalopathy with focal neurological findings and impaired awareness, mus- cle weakness, lactic acidosis, diabetes, cardiac disease, and gas- trointestinal dysmotility [2].
The heart has a high metabolic demand, and mitochondrial function is a key determinant of myocardial performance [4].
A recent meta-analysis showed that patients with MELAS syndrome have the highest prevalence of electrocardiographic and echocardiographic abnormalities compared to other mitochondrial diseases [5]. Cardiac involvement is reported in more than 50% of patients with the 3243A>G mutation. Hypertrophic remodeling is the most common early phenotype, possibly evolving in DCM at a later stage and resulting in significant heart rhythm disorders in many patients [6,7]. In the early stages of the disease, myocardial hypertrophy occurs as an adaptive response to mitochondrial energy deficits. However, as mitochondrial dysfunction worsens, the heart’s ability to generate energy becomes further compromised, leading to myocardial fibrosis, loss of contractile function, and eventual progression to DCM.
An intriguing aspect of MELAS-related cardiomyopathy (MELAS-CM) is the temporal heterogeneity of its clinical on- set. While cardiac involvement is rarely documented in infancy, most patients develop cardiac manifestations later in life [5-7].
Pediatric-onset MELAS-CM, as seen in this case, has been rarely reported in the literature [8]. It is typically associated with more severe systemic disease and a worse prognosis. In contrast, adult-onset MELAS-CM tends to be more stable and is less frequently implicated in life-threatening cardiac events [7].
Given the high prevalence of cardiac involvement in MELAS syndrome, careful and regular cardiac follow-up is mandatory in all patients. Serial echocardiographic assessments, electro- cardiograms, and in selected cases cardiac Magnetic Resonance Imaging (MRI) can help monitor disease progression and guide therapeutic interventions. Early initiation of cardioprotective medications, such as beta-blockers, ACE inhibitors, and aldos- terone antagonists, may help delay disease progression and im- prove patient outcomes.
However, the management of MELAS-CM remains an area of unmet clinical need. Unlike other forms of cardiomyopathy, where heart transplantation (Htx) is a viable option, the multi- systemic nature of MELAS syndrome often precludes transplan- tation in affected individuals. Neurologic impairment, recurrent stroke-like episodes, chronic kidney disease, and wasting my- opathy present significant barriers to Htx eligibility in both pedi- atric and adult patients [9]. Htx is controversial and has rarely been performed with conflicting results. Recently, a success- ful simultaneous heart-kidney transplantation in a 30-year-old male patient with MELAS syndrome has been described [10].
Our case highlights the early and aggressive cardiac involve- ment observed in a pediatric patient with MELAS syndrome, emphasizing the need for close cardiac monitoring in these pa- tients. Pediatric-onset MELAS-CM, as seen in this case, has been rarely reported in the literature. It is typically associated with more severe systemic disease and a worse prognosis.
Multidisciplinary collaboration among metabolic specialists, neurologists, and cardiologists is essential for optimizing care of MELAS patients. The multisystemic nature of MELAS syndrome complicates therapeutic decision-making, particularly in cases of severe CM where Htx may be considered. Additionally, the unpredictability of organ and tissue involvement due to hetero- plasmy further complicates clinical management, making treat- ment decisions particularly challenging.
Future research is needed to explore novel therapeutic strat- egies aimed at preserving mitochondrial function and mitigat- ing the cardiac manifestations of this devastating disorder.
Acknowledgments: We would like to thank AISMME (Associazione Italiana Supporto Malattie Metaboliche Ereditarie/ Italian Association Inherited Metabolic Diseases Support) for funding grants and continuous support to clinical activities of Inherited Metabolic Diseases Unit of Department of Pediatrics at the University Hospital of Verona (Italy).
Some Authors of this publication are members of the Euro- pean reference Network for Rare Hereditary Metabolic Disor- ders (MetabERN) project ID N 739543.
We want to dedicate this work to our little patient and his parents and we would like to deeply thank them for permission they gave us to publish all the data making so possible the col- laboration between clinical centers and various professionals in the hope to strength multidisciplinary efforts and to increase the knowledge and expertise in mitochondrial disorders.
Informed consent statement and ethical approvals: The lat- est revision of the Helsinki declaration as well as the Oviedo declaration were the basis for the ethical conduct of the study. The study protocol was designed and conducted to ensure ad- herence to the principles and procedures of good clinical prac- tice and to comply with the Italian laws. Written informed con- sent for publication of the clinical details was obtained from the parents of patient.
Competing interest statement: None of the authors have competing interests to declare. None of the authors accepted any reimbursements, fees or funds from any organization that may in any way gain or lose financially from the results of this study.
Funding: This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author contribution: Conceptualization, GM, EM, AB; meth- odology, AB; investigation, GR, AMP, GG, AD; data curation, GM, AMP, GG, AD; writing-original draft preparation, GM, EM; writ- ing-review and editing, RF, NV, LR, AMP, AS, AB. All authors have read and agreed to the published version of the manuscript.
Abbreviations: CM: Cardiomyopathy; DCM: Dilated Car- diomyopathy; E/E’ Ratio: The Ratio Of Mitral Peak Velocity Of Early Filling (E) To Early Diastolic Mitral Annular Velocity (E’); HCM: Hypertrophic Cardiomyopathy; Htx: Heart Transplanta- tion; IVS: Interventricular Septum; LVEDD: Left Ventricular End Diastolic Diameter; LVESD: Left Ventricular End Systolic Diam- eter; LVPW: Left Ventricular Posterior Wall; LVFS: Left Ventricu- lar Fractional Shortening; LVEDV: Left Ventricular End-Diastolic Volume; LVESV: Left Ventricular End-Systolic Volume LVEF: Left Ventricular Ejection Fraction; MELAS: Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, And Stroke; Mtdna: Mitochon- drial DNA.
