-
Email info@annpnc.org
-
Address 848 N. Rainbow Blvd. #5486 Las Vegas, NV 89107, USA
1Junior Resident, Department of Pediatrics, TNMC and BYL Nair Charitable Hospital, India.
2Assistant Professor, Department of Pediatrics, TNMC and BYL Nair Charitable Hospital, India
3Additional Professor, Department of Pediatrics, TNMC and BYL Nair Charitable Hospital, India.
4Head of Department, Department of Pediatrics, TNMC and BYL Nair Charitable Hospital, India.
*Corresponding author: Gulrej Shaikh
Assistant Professor, Department of Pediatrics, TNMC
and B.Y.L Nair Charitable Hospital, Mumbai 400008, India.
Email ID: gulrej24@gmail.com
Received: Jan 29, 2025
Accepted: Mar 14, 2025
Published Online: Mar 21, 2025
Journal: Annals of Pediatrics and Neonatal Care
Copyright: Shaikh G et al. © All rights are reserved
Citation: Bhavana MP, Shaik G, Kondekar S, Rathi S. A rare case of spinal plexiform neurofibromas in a 9-year-old Indian child with Neurofibromatosis-Noonan syndrome: Clinical and neuroradiological insights. Ann Pediatr Neonatal Care. 2025; 1(1): 1001.
Neurofibromatosis-Noonan Syndrome (NFNS) is a rare RASopathy resulting from a variant NF1 gene mutation, characterized by a combination of features from both Neurofibromatosis type 1 (NF1) and Noonan Syndrome (NS). We present a case of a 9-year-old boy who exhibited learning difficulties, multiple café-au-lait macules, and genitourinary concerns. Child did not satisfy the standard NIH criteria for NF1. Magnetic Resonance Imaging (MRI) revealed Focal Areas of Signal Intensities (FASI) in the brain and multiple plexiform neurofibromas in the nerve roots of dorsolumbar spine. Genetic analysis confirmed a heterozygous pathogenic variant in the NF1 gene, consistent with NFNS. This is the first such case of unusual presentation of spinal plexiform neurofibromas in a child with NFNS, expanding the phenotypic spectrum of this rare syndrome.
Keywords: Neurofibromatosis-Noonan syndrome; RASopathy; Spinal plexiform neurofibroma; Focal areas of signal intensities.
Neurofibromatosis-Noonan Syndrome (NFNS) is a rare RASopathy that presents with overlapping clinical features of Neurofibromatosis type 1 (NF1) and Noonan Syndrome (NS). Approximately 25% of NF1 patients exhibit features suggestive of NS, linking these conditions through mutations in the RAS/ MAPK pathway. NFNS is a rare disorder primarily caused by mutations in the NF1 gene or PTPN11 genes [1]. We present a rare case of NFNS with an unusual manifestation of spinal plexiform neurofibroma in a 9-year-old boy with learning disabilities.
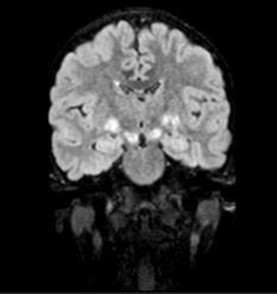
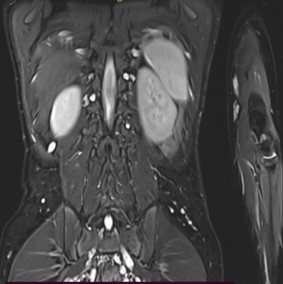
A 9-year-old male presented with a history of learning difficulties and poor scholastic performance since age six. Diagnosed with a specific learning disability-dyslexia, and Attention Deficit Hyperactivity Disorder (ADHD), he also experienced intermittent abdominal pain, enuresis, and encopresis. His development was appropriate for age. He had a borderline IQ of 75 (Wechsler Intelligence Scale for Children). He was operated for exotropia at the age six. There was no family history of neurodevelopmental or neurocutaneous disorders. Physical examination revealed short stature (Height: 128 cm, at 10th percentile), weight of 25 kg (25th-50th percentile), and a head circumference of 52 cm (50th-90th percentile). Multiple café-au-lait macules (more than 15, with only four larger than 5 mm) were noted. Dysmorphic facial features, including a flat facial profile, lowset ears, and dental malocclusion, were observed. Neurological examination was remarkable for hyperesthesia over the C6T1 dermatomes bilaterally with no other neurological deficits. Given the incomplete fulfillment of NIH criteria for café au lait macules in Neurofibromatosis Type 1, additional investigations were pursued. Slit lamp examination revealed a single iris Lisch nodule in the left eye. Both audiological and orthopedic assessments were normal. Abdominal ultrasonography and echocardiography were normal. Brain MRI identified ‘Focal Areas of Signal Intensities (FASI)’ in the bilateral globus pallidus, crus cerebri, and dentate nuclei, consistent with NF1 (Figure 1). Spinal MRI with contrast, disclosed multiple bilateral paravertebral enhancing nodular lesions in the dorso-lumbar spine and sacral canal with the largest at L4-L5 level measuring 2.6X2.4X3.2 cm in the left paravertebral region along with mild extension into the L3-L4 and L4-L5 neural foramina, consistent with plexiform neurofibromas (Figure 2). Genetic testing confirmed a heterozygous c.499del (p. Cys167ValfsTer11) pathogenic variant in the NF1 gene, affirming the diagnosis of Neurofibromatosis Noonan’s Syndrome (NFNS). The patient was initiated on methylphenidate in addition to behavioral interventions and individualized educational plan for the specific learning disability and ADHD. Neurosurgical consult for spinal plexiform neurofibromas, recommended conservative management with a trial of the novel drug, Selumetinib, a MEK inhibitor, for the spinal plexiform neurofibroma. The family received counseling on the chronic nature of the condition, the importance of close follow up, behavioral interventions for the comorbidities and the potential need for surgical intervention in the future if the spinal plexiform neurofibromas increase in size or develop a malignant transformation.
The RASopathies, including NF1 and NS, constitute a group of genetic syndromes caused by mutations affecting the RAS/ MAPK signaling pathway, with an estimated prevalence of 1 in 1,000 live births. These disorders include neurofibromatosis type 1, Noonan syndrome, Noonan syndrome with multiple lentigines, Capillary malformation–arteriovenous malformation syndrome, Costello syndrome, Cardio-facial-cutaneous syndrome, and Legius syndrome. All these disorders share a common pathophysiology i.e. dysregulation of the RAS/MAPK pathway, leading to overlap of clinical manifestations which includes cranio-facial dysmorphism, malformations of the heart, eye, brain, neurocognitive impairment and increased carcinogenic risk [2].
NFNS, first described in 1985, where four unrelated patients were initially diagnosed as NF1 but exhibited features of both NF1 and Noonan Syndrome (NS). These patients were labelled as NFNS. Among NFNS patients, several patients were found to have mutations in both PTPN11 and NF1 genes. However, majority of NFNS patients, have been reported to have isolated NF1 mutations, without any detectable PTPN11 mutation. Thus, NFNS syndrome has been attributed to NF1 mutations by most authors, as in our case [3,4].
NF1 is clinically defined by neurofibromas, café-au-lait macules (6 in number>5 mm in size for pre-pubertal age group and >15 mm in size for post-pubertal age), axillary or inguinal freckling, Lisch nodules in the Iris (>2 in no.), optic nerve gliomas, osseous lesions and history of an affected first degree relative. Presence of two or more out of 6 of “the United States National Institutes of Health (NIH)” criteria of the updated version (2021) is consistent with the diagnosis of this disease [5]. Although clinical diagnosis relies on the NIH criteria, our case underscores the importance of neuroimaging and molecular diagnostics, particularly in atypical presentations that do not satisfy the NIH criteria for Neurofibromatosis type 1. Focal Areas of Signal Intensities (FASI) have been reported to be associated with clinical and genetically proven NF1 cases. Basal ganglia are the most frequent localization of these lesions as well as cerebellum and brainstem [6]. Spinal plexiform neurofibromas, though rare, can cause significant morbidity due to their potential for nerve root compression and subsequent neurological deficits. The introduction of targeted therapies, such as MEK inhibitors like Selumetinib, offers promising alternatives to surgical management, potentially improving clinical outcomes in patients with complex RASopathies [7].
Our patient’s presentation with learning disabilities, incomplete NF1 features complemented by neuroradiological findings of spinal plexiform neurofibromas and associated Focal Areas of Signal Intensities (FASI) in the brain, are characteristic of NF1; however, this combination of features has not yet been reported till date in a child with Neurofibromatosis-Noonan Syndrome. Molecular confirmation of the NF1 mutation further confirmed the diagnosis, highlighting the utility of genetic testing in such complex phenotypic presentation.
Our findings suggest a need to reevaluate the diagnostic criteria for NF1 to include cases with incomplete or atypical clinical presentations. This change could facilitate early diagnosis and comprehensive management of the affected children with NF1, NFNS and their phenotypic variants. Early clinical suspicion and neuroradiological evaluation can unmask clinically dormant spinal plexiform neurofibromas, allowing for timely therapeutic interventions. This case exemplifies the critical role of neuroradiology and molecular diagnostics in identifying rare syndromic associations as NFNS.
